Techniques
Systems & Synthetic Biology
Engineering gene constructs to precisely control bacterial behavior, kinetics, and species interactions
Computational biology
Leveraging next-gen sequencing data and bioinformatics to gain mechanistic insights
Single cell quantification
Employing cutting-edge microscopy, flow cytometry, biosensors, and quantitative assays for single-cell measurements
Data Science & ML
Analyzing data with ML & AI techniques to predict findings and drive new discovery
Mathematical modeling
Building mechanistic models of gene and cellular network interactions to predict and optimize system behaviors
Biomolecular engineering
Applying engineering principles to design and evolve genetic components with desired properties
Research Areas
Antibiotic Resistance

The rapid emergence of antibiotic-resistant pathogens presents a significant threat to global public health, exacerbated by a stagnating drug discovery pipeline.
Our work focuses on uncovering the metabolic pathways and genetic mechanisms underlying resistance evolution.
We are leveraging these insights to develop innovative approaches for enhancing the efficacy of existing antibiotics, including diagnostic tools and therapeutic interventions that could restore their potency against resistant strains
AJ Lopatkin, 2021,
AJ Lopatkin, 2019,
SV Aduru, 2024,
A Palomino, 2022.
Horizontal Gene Transfer
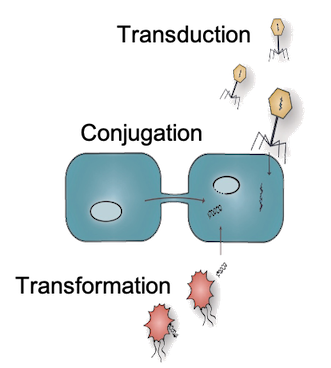
HGT is a key mechanism by which bacteria exchange genetic material, spreading traits like antibiotic resistance and stabilizing microbial communities.
Despite its importance, the fundamental kinetic principles governing HGT remain poorly understood.
Our lab uses precise kinetic measurements, single-cell quantification, and AI/ML-driven genomic discovery to uncover these dynamics.
We are uncovering these dynamics through precise kinetic measurements, single-cell quantification, and AI/ML-driven genomic discovery, and applying this knowledge to engineer microbial systems with controlled and predictable genetic exchange properties
AJ Lopatkin, 2017,
AJ Lopatkin, 2016,
H Prensky, 2021,
M Ahmad, 2023.
Microbial Community Engineering

Microbial communities, or microbiomes, play vital roles in maintaining health and stability across diverse ecosystems, from the human body to environmental niches.
Our lab aims to understand the fundamental design principles that govern microbial community stability and function.
Ultimately, we aim to design and engineer microbial populations with precise, tailored functionalities that address challenges in health, agriculture, and industrial applications.
S Huang, 2015,
T Wang, 2022,
F Wu, 2019.
Environmental Applications

Pollution from industrial and agricultural activities has led to the accumulation of harmful chemicals in ecosystems worldwide. Microbes offer a sustainable, green solution for breaking down these pollutants.
Our lab studies the poorly understood dynamics of microbial degradation processes and the risks of unintended genetic exchange in these settings.
We focus on optimizing microbial systems for the removal of specific pollutants (e.g., alkanes) while developing predictive models to assess the ecological risks associated with genetic contamination
J Moralez, 2021,
ME Schoen, 2021,
SC Williams, 2021.